Michelle Z. Gurvitz
Người dịch: BS Ngô Thị Kim Ánh
Một bệnh nhân nữ 23 tuổi đến khám tại phòng khám bệnh tim bẩm sinh người lớn sau 8 năm không theo dõi. Triệu chứng hiện tại của bệnh nhân là mệt nhẹ khi gắng sức. Bệnh nhân có bệnh sử hẹp eo động mạch chủ và thông liên thất đã được phẫu thuật từ sơ sinh. Bệnh nhân được phẫu thuật sửa chữa hẹp eo động mạch chủ bằng miếng vá và phải phẫu thuật lần 2 lúc 10 tuổi do tái hẹp. Ở lần phẫu thuật sau, bệnh nhân cũng được tạo hình động mạch chủ bằng miếng vá.
Khi thăm khám, bệnh nhân có mạch chi trên trái và mạch đùi phải yếu, huyết áp tâm thu tay phải cao hơn chân phải 32 mmHg. Hình ảnh siêu âm của cung động mạch chủ hạn chế nhưng ghi nhận có chênh áp tồn lưu 41 mmHg, phổ Doppler động mạch chủ dưới có đỉnh tâm thu tù và phổ kéo dài trong thì tâm trương. Chức năng tâm thu thất trái giảm nhẹ với phân suất tống máu 45- 50%. Hình ảnh MRI cho thấy hẹp eo động mạch chủ tồn lưu và phình tại miếng vá eo động mạch chủ.
Mở đầu
Các khiếm khuyết bẩm sinh làm ảnh hưởng đến tim và các mạch máu lớn xuất phát từ tim, đó là động mạch chủ và động mạch phổi. Chương này tập trung vào những bất thường của động mạch chủ. Các chủ đề được bàn luận bao gồm hẹp eo động mạch chủ và các bệnh lý liên quan đến bất thường mô liên kết của động mạch chủ. Chương này sẽ nói về chẩn đoán, biểu hiện lâm sàng, điều trị và theo dõi bệnh, tập trung vào những bệnh ở người lớn và những di chứng lâu dài sau sửa chữa hoặc điều tri bệnh,
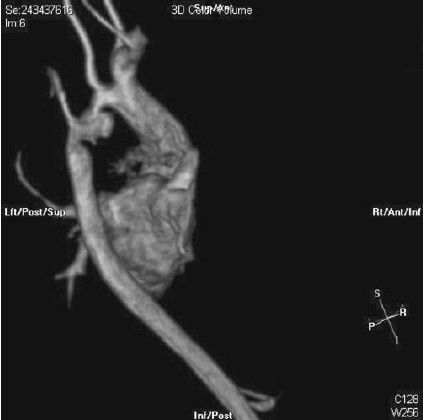
Hình 8.1: Hình ảnh MRI cho thấy hẹp eo động mạch chủ tồn lưu và phình sau phẫu thuật tạo hình động mạch chủ.
Phôi thai học và sự phát triển của động mạch chủ
Động mạch chủ là một cấu trúc phức tạp cả về nguồn gốc phôi thai và cấu trúc mô học. Về mặt phôi thai học, động mạch chủ là sự gắn kết của nhiều cấu trúc như thân chung động mạch, túi động mạch chủ, cung cánh tay 3 -4 và động mạch chủ lưng. Ban đầu là cấu trúc đôi, sau đó thoái triển dần thành một cung động mạch chủ duy nhất. Thân chung động mạch và túi động mạch chủ tạo thành động mạch chủ lên, cung thứ 3 tạo thành động mạch cảnh và động mạch dưới đòn, cung thứ 4 giúp hoàn thiện động mạch chủ ngang, sau đó động mạch chủ xuống được hình thành từ động mạch chủ lưng phôi thai. Ống động mạch hình thành từ cung thứ 6, ngay sau động mạch dưới đòn trái [1].
Mất điều hòa của bất kỳ thành phần nào cũng có thể dẫn đến dị dạng động mạch chủ hoặc làm thay đổi vị trí bên trái bình thường của động mạch chủ. Ví dụ như động mạch dưới đòn phải xuất phát bất thường từ động mạch chủ xuống và cung động mạch chủ nằm bên phải là do sự thoái triển bất thường của các cấu trúc bên trái trong thời kỳ phôi thai. Cung động mạch chủ soi gương nằm bên phải hiếm khi kết hợp với hẹp eo động mạch chủ nhưng thường đi kèm với các bất thường bẩm sinh tim khác như tứ chứng Fallot [2].
Về mặt mô học, động mạch chủ là cấu trúc 3 lớp bao gồm: nội mạc, trung mạc và ngoại mạc. Nội mạc là lớp trong cùng và được bao phủ bởi lớp tế bào nội mạc. Trung mạc là một lớp cơ bao gồm các tế bào cơ trơn, fibrillin, elastin và collagen, xen kẽ với chất gian bào. Ngoại mạc là lớp ngoài cùng của động mạch chủ [3].
HẸP EO ĐỘNG MẠCH CHỦ
Định nghĩa – tần suất
Hẹp eo động mạch chủ là tình trạng hẹp động mạch chủ mà gây giảm dòng máu đến các mạch máu và các cơ quan ở xa kèm tăng huyết áp phần gần.
Độ nặng của hẹp eo động mạch chủ thay đổi và thường gặp nhất ở đoạn gần động mạch chủ ngực, ngay sau chỗ xuất phát động mạch dưới đòn trái, tại vị trí ống động mạch. Hiếm hơn, hẹp động mạch chủ có thể xảy ra ở động mạch chủ bụng hoặc có thể gặp thiểu sản cung động mạch chủ lan tỏa.
Hẹp eo động mạch chủ là một bệnh tim bẩm sinh tương đối thường gặp, chiếm 6-8% bệnh tim bẩm sinh [4,6], tỷ lệ nam/nữ ít nhất là 1.5 – 2. Mặc dù phần lớn các trường hợp không có di truyền, nhưng các bài báo gần đây xác nhận có thể có sự kết hợp về gen với các tổn thương tắc nghẽn bên trái, bao gồm hẹp eo động mạch chủ [7,8]. Hẹp eo động mạch chủ cũng kết hợp với bệnh lý về gen khác như hội chứng Turner [9,10].
Hẹp eo động mạch chủ có thể đơn độc hoặc kết hợp với các bất thường bẩm sinh tim khác, thường gặp nhất là van động mạch chủ 2 mảnh ( 20-40% bệnh nhân)[4,11]. Các tổn thương kết hợp khác là thông liên thất, tồn tại ống động mạch, hẹp van động mạch chủ và bất thường van 2 lá [12]. Trong vài trường hợp hẹp eo động mạch chủ kéo dài, tuần hoàn bàng hệ bù trừ phát triển từ động mạch ngực trong và động mạch gian sườn, đi tắt qua chổ hẹp để cung cấp máu cho chi dưới.
Mặc dù bệnh sinh chính xác của hẹp eo động mạch chủ không rõ ràng nhưng có 2 giả thiết chiếm ưu thế. Giả thiết thứ nhất cho rằng hẹp eo động mạch chủ là một hiện tượng liên quan đến dòng máu. Hạn chế dòng máu động mạch chủ trong thời kỳ phôi thai dẫn đến sự phát triển bất thường của cung động mạch chủ [12,13], Giả thiết này phù hơp với dấu hiệu thường gặp là van động mạch chủ 2 mảnh và thông liên thất. Giả thiết thứ hai cho rằng, hẹp eo động mạch chủ là hậu quả của mô động mạch chủ bất thường, một phần của mô này lan từ ống động mạch đến động mạch chủ. Khi ống động mạch đóng , thì động mạch chủ bị siết lại ở nhiều mức độ khác nhau. Mô học của vùng hẹp cho thấy có hiện tượng dày, đứt đoạn lớp trung mạc và tăng sinh lớp nội mạc. Các nghiên cứu gần đây cũng cho thấy bất thường mô ở vùng trước và sau chỗ hẹp: xáo trộn lớp collagen, mất cơ trơn, tăng độ cứng động mạch chủ [14,16]. Những bằng chứng gần đây phù hợp hơn với bất thường mô học, tuy nhiên, ảnh hưởng huyết động học của giảm dòng máu cũng đóng vai trò trong cơ chế bệnh sinh.
Biểu hiện lâm sàng và chẩn đoán
Mặc dù hẹp eo động mạch chủ là bất thường bẩm sinh động mạch chủ nhưng nhiều bệnh nhân không được chẩn đoán cho đến khi trưởng thành. Triệu chứng lâm sàng khi chẩn đoán phụ thuộc vào tuổi bệnh nhân. Hầu hết các trường hợp hẹp eo động mạch chủ nặng có biểu hiện trong thời kỳ sơ sinh vì dòng máu hệ thống phụ thuộc vào dòng máu qua ống động mạch. Chẩn đoán có thể khó khăn nếu còn ống động mạch, tuy nhiên, vài trẻ có biểu hiện tím phân biệt với chân tím nhiều hơn tay. Những trẻ này sẽ suy tim nặng hoặc sốc tim khi ống động mạch đóng. Rất may, những trường hợp này có thể điều trị bằng prostaglandin truyền tĩnh mạch nhằm duy trì mở ống động mạch, bảo tồn dòng máu hệ thống cho đến khi có thể phẫu thuật.
Hẹp eo động mạch chủ thể nhẹ biểu hiện trễ ở trẻ lớn hoặc người lớn. Mặc dù phần lớn bệnh nhân không triệu chứng, một số ít bệnh nhân có triệu chứng lạnh chân, mỏi chân hoặc cơn đau cách hồi khi gắng sức. Thường hơn. hẹp eo động mạch chủ được chẩn đoán do tăng huyết áp kháng trị, do phát hiện âm thổi tại chỗ hẹp, hoặc tổn thương phối hợp khác.
Thăm khám tim mạch toàn diện rất quan trọng trong chẩn đoán hẹp eo động mạch chủ. Trong trường hợp bình thường, huyết áp chi dưới cao hơn chi trên. Ở bệnh nhân hẹp eo động mạch chủ huyết áp chân thấp hơn tay phải. Khi có tuần hoàn bàng hệ lớn hoặc động mạch dưới đòn phải xuất phát bất thường, sự khác biệt huyết áp không rõ ràng gây khó khăn cho chẩn đoán. Nên khám kỹ mạch quay, mạch cánh tay và mạch đùi về biên độ và thời gian. Tương tự như việc giảm huyết áp chi dưới ở bệnh nhân hẹp eo động mạch chủ, mạch đùi đập yếu hơn và đập sau động mạch tay phải. Mạch và huyết áp tay trái thay đổi tùy thuộc vào vị trí chỗ hẹp so với động mạch dưới đòn trái.
Khám tim cho thấy tăng xung động thất trái do phì đại thất trái. Âm thổi của hẹp eo động mạch chủ là âm thổi tâm thu, lan qua tâm trương ngắn, nghe rõ nhất ở vùng quanh cột sống trái, gần xương vai trái. Nếu có nhiều tuần hoàn bàng hệ quanh vị trí hẹp eo, có thể nghe được âm thổi liên tục sau lưng. Nếu bệnh nhân có tổn thương phối hợp thì có thể nghe được các âm thổi khác, thường gặp nhất là âm thổi tâm thu và tâm trương của hẹp hoặc hở van động mạch chủ do van động mạch chủ 2 mảnh. Van động mạch chủ 2 mảnh cũng có click tâm thu.
Chẩn đoán hình ảnh
Khi nghi ngờ hẹp eo động mạch chủ, nhiều trắc nghiệm có thể giúp chẩn đoán xác định. Các khảo sát hình ảnh cắt ngang kỹ thuật cao cung cấp thông tin giải phẫu rất tốt nhưng không phải lúc nào cũng có sẵn. Trong khi đó, các phương tiện chẩn đoán thông thường hơn như X quang và điện tâm đồ cũng hữu dụng. Hình ảnh X quang ở bệnh nhân hẹp eo động mạch chủ lớn tuổi có thể thấy tim hơi lớn, dãn nhẹ động mạch chủ lên, phần đầu động mạch chủ lên nhô, hẹp đoạn xa động mạch chủ ngang và dãn động mạch chủ xuống. Những đoạn dãn này tạo thành hình ảnh “số 3” kinh điển, hình ảnh này không thường gặp trên nhiều phim X quang. Cũng có thể thấy hình ảnh “khuyết xương sườn” do sự bào mòn bờ dưới của các xương sườn vùng giữa ngực phía sau, nguyên nhân là do hẹp eo động mạch chủ làm lớn các động mạch liên sườn.(hình 8.2)
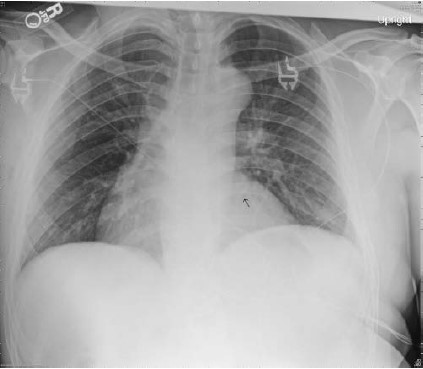
Hình 8.2: Hình ảnh X quang của một bệnh nhân người lớn có hẹp eo động mạch chủ chưa sửa chữa. Ghi nhận bóng cung động mạch chủ dãn do dãn động mạch chủ phía trước và sau chỗ hẹp. Mũi tên đen chỉ dấu mòn xương sườn.
Điện tâm đồ trong hẹp eo động mạch chủ tương đối bình thường, ngoại trừ dấu hiệu lớn các buồng tim. Sơ sinh có hình ảnh lớn thất phải đơn thuần, trong khi người lớn có hình ảnh lớn thất trái.
Bên cạnh X quang, siêu âm 2 chiều và Doppler là phương tiện chẩn đoán hình ảnh thông dụng và dễ thực hiện nhất. Siêu âm cung cấp các thông tin về giải phẫu và huyết động rất tốt, đồng thời siêu âm cũng giúp phát hiện các bất thường tim bẩm sinh kết hợp. Cung động mạch chủ thấy rõ nhất ở mặt cắt trên hõm ức và trục dọc cạnh ức cao. Hình ảnh chỗ hẹp có thể thấy được trên 2D. Phổ Doppler có vận tốc cao thì tâm thu và phổ giảm nhanh thì tâm trương tại chỗ hẹp ( hình 8.3a). Vận tốc tối đa và trung bình có thể giúp xác định độ nặng của hẹp eo động mạch chủ nhưng có thể đánh giá quá mức khi có hẹp đoạn dài hoặc tăng độ cứng thành mạch. Ở mặt cắt dưới sườn, phổ động mạch chủ bụng có đỉnh tâm thu tù và phổ giảm nhanh thì tâm trương ( hình.8.3b)
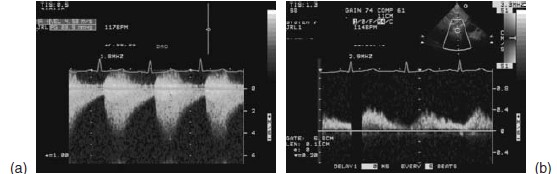
Hình 8.3: Hình ảnh siêu âm gợi ý hẹp eo động mạch chủ
a) Phổ Doppler liên tục cho thấy tăng vận tốc đỉnh với phổ kéo dài thì tâm trương qua chỗ hẹp.
b) Dòng Doppler có đỉnh tù của động mạch chủ bụng.
Ở vài bệnh nhân, đánh giá giải phẫu bằng siêu âm tim gặp khó khăn do cửa sổ siêu âm kém, khi đó MRI và CT là phương tiện chẩn đoán hình ảnh hữu dụng để xác định cấu trúc giải phẫu động mạch chủ và chẩn đoán hẹp eo động mạch chủ [17-19]. Hai phương tiện chẩn đoán hình ảnh này đều cho nhiều hình ảnh 2D cũng như tái tạo 3D của động mạch chủ ( hình 8.4 ). Tuy nhiên, vài trung tâm không có dụng cụ và kinh nghiệm để khảo sát tim. Mặc dù MRI thường được lựa chọn vì bệnh nhân không phải tiếp xúc với tia xạ và có thể cung cấp thông tin về huyết động học, nhưng hiện nay nhiều bệnh nhân có chống chỉ định với MRI như bệnh nhân có máy tạo nhịp hoặc đặt dụng cụ bằng kim loại. CT scan có thể thực hiện được trên bệnh nhân có dụng cụ bằng kim loại nhưng bệnh nhân phải tiếp xúc với tia X. Cuối cùng, việc lựa chọn phương tiện chẩn đoán hình ảnh tùy thuộc từng bệnh nhân và kinh nghiệm về tim bẩm sinh của người thực hiện.

Hình 8.4: Hình ảnh CT scan của một bệnh nhân người lớn có hẹp eo động mạch chủ chưa sửa chữa. Ghi nhận dãn động mạch chủ trước và sau đoạn hẹp. Mũi tên chỉ các tuần hoàn bàng hệ.
Thông tim cho biết những thông tin quan trọng liên quan đến hẹp eo động mạch chủ, chênh áp ngang chỗ hẹp và các thông số huyết động học xâm nhập khác. Tuy nhiên, hẹp eo động mạch chủ có thể được chẩn đoán xác định bằng các phương tiện chẩn đoán hình ảnh không xâm nhập, thông tim nên được dùng trong can thiệp điều trị, thường chỉ định khi độ chênh áp tối đa trên 20 mmHg.
Điều trị
Khi hẹp eo động mạch chủ được chẩn đoán, phẫu thuật và can thiệp bằng thông tim là hai lựa chọn cần được xem xét nhằm giải phóng chỗ hẹp. Cần sửa chữa hẹp eo động mạch chủ ngay thời điểm chẩn đoán. Các nghiên cứu theo dõi lâu dài cho thấy tỷ lệ tử vong tăng ở những bệnh nhân không được điều trị hoặc điều trị trễ [20-28]. Một nghiên cứu cho thấy bệnh nhân không điều trị có tỷ lệ tử vong sớm cao với tuổi tử vong trung bình ở khoảng 40 tuổi và 75% chết trước 46 tuổi [20]. Các nguyên nhân tử vong thường gặp trong nghiên cứu này và các nghiên cứu tử thiết khác là suy tim sung huyết, vỡ động mạch chủ, viêm nội tâm mạc nhiễm trùng và xuất huyết nội sọ [20-22]. Thậm chí cho tới những năm gần đây, những bệnh nhân được phẫu thuật vẫn có tuổi thọ giới hạn, nhất là khi phẫu thuật được thực hiện khi trẻ đã lớn [23-28]. Những bệnh nhân này có tỷ lệ mắc bệnh tăng huyết áp cao và các nguyên nhân tử vong hàng đầu là bệnh mạch vành, đột tử và suy tim [23,26].
Chỉ định can thiệp hẹp eo động mạch chủ là chênh áp lớn hơn 20 mmHg (xác định bằng đo huyết áp tâm thu, siêu âm tim hoặc thông tim), có bằng chứng tăng huyết áp, rối loạn chức năng thất trái, hoặc suy tim sung huyết. Sự phát triển tuần hoàn bàng hệ cũng là một chỉ định can thiệp, vì trong trường hợp này chênh áp qua chỗ hẹp sẽ thấp hơn thực tế. Hiện nay, phẫu thuật và các kỹ thuật thông tim đều có thể lựa chọn để can thiệp. Trong nhiều tình huống, lựa chọn cách can thiệp vẫn là một vấn đề còn bàn cãi.
Phẫu thuật
Hẹp eo động mạch chủ là một trong những bệnh tim bẩm sinh được phẫu thuật đầu tiên. Vào những năm giữa thập niên 40, bác sĩ Crooford và Nylin ở Thụy Điển và bác sĩ Grooss ở Mỹ đã phẫu thuật thành công trường hợp hẹp eo động mạch chủ đầu tiên. Sau đó, các kỹ thuật phẫu thuật đã được thay đổi và cải tiến. Các phương pháp phẫu thuật hiện nay bao gồm:
- Nối tận-tận ( hoặc nối tận-tận có mở rộng). Đoạn động mạch chủ bệnh được cắt bỏ, đầu gần và đầu xa được khâu nối lại với nhau.
- Sửa chữa bằng miếng vá động mạch dưới đòn. Động mạch dưới đòn trái được cắt ngang và phần gần được dùng để mở rộng đoạn động mạch chủ bị hẹp.
- Bắc cầu qua chỗ hẹp. Một ống ghép được dùng làm cầu nối qua chỗ hẹp
- Tạo hình động mạch chủ bằng miếng vá. Một miếng vá được khâu vào động mạch chủ để làm mở rộng đoạn hẹp.
Lựa chọn phương pháp phẫu thuật phụ thuộc vào tuổi và kích thước bệnh nhân, giải phẫu của cung động mạch chủ và chỗ hẹp, các tổn thương phối hợp. Ở trẻ sơ sinh và trẻ nhỏ, phẫu thuật nối tận-tận thường được sử dụng. Phương pháp sửa chữa bằng miếng vá (nắp) động mạch dưới đòn được dùng trong những tổn thương đoạn dài. Ở trẻ lớn, trẻ vị thành niên và người lớn, thường thực hiện phẫu thuật nối tận-tận. Tuy nhiên đối với tổn thương dài và phức tạp thì cần phẫu thuật bắc cầu. Mặc dù kỹ thuật sửa chữa bằng miếng vá (nắp) động mạch dưới đòn thường được lựa chọn cho trẻ sơ sinh, nhưng vài báo cáo ghi nhận có sự giảm phát triển và hội chứng đau thần kinh ở tay trái [31]. Hiện nay, tạo hình động mạch chủ bằng miếng vá hiếm khi thực hiện vì nó có thể phát triển thành phình và vỡ động mạch chủ sau một thời gian và cũng có nguy cơ tái hẹp tương đối cao.
Tỷ lệ tử vong của phẫu thuật sửa chữa hẹp eo động mạch chủ thấp khi được thực hiện bởi phẫu thuật viên tim bẩm sinh có kinh nghiệm. Tần suất biến chứng tăng ở những bệnh nhân lớn và bệnh nhân có tổn thương tim bẩm sinh kết hợp. Do động mạch chủ nằm gần các cấu trúc quan trọng khác nên các biến chứng có thể xảy ra trong lúc phẫu thuật. Tổn thương dây thần kinh thanh quản quặt ngược hoặc thần kinh hoành dẫn đến liệt dây thanh âm hoặc liệt cơ hoành. Một trong những biến chứng nặng nhưng không thường gặp là tổn thương cột sống và gây liệt, thường gặp ở người lớn và trẻ em hơn là trẻ sơ sinh, có thể do giảm tưới máu tủy sống và thiếu máu khi kẹp động mạch chủ ở bệnh nhân có tuần hoàn bàng hệ kém [32-34]. Chú ý hơn đến tưới máu đoạn xa, giảm thời gian kẹp động mạch chủ hoặc tim phổi nhân tạo một phần giúp làm giảm nguy cơ tổn thương tủy sống.
Một vấn đề thường gặp sau mổ là tăng huyết áp nghịch thường. Tăng huyết áp do tăng phóng thích catechol và hoạt tính giao cảm qua trung gian thụ thể hóa học, cũng như những thay đổi trong trục renin-aldosterone [35,36]. Ở giai đoạn đầu sau mổ, thường phải dùng thuốc hạ áp tích cực đường tĩnh mạch, nhưng bệnh nhân thường có thể chuyển sang đường uống sau vài ngày khi phản ứng tăng huyết áp giảm. Một biến chứng ít gặp hơn là “hội chứng sau cắt hẹp eo” bao gồm: đau bụng, căng bụng và nôn ói. Hội chứng này liên quan đến tăng huyết áp và tăng ḍòng máu đến mạch máu mạc treo sau phẫu thuật. Có thể pḥòng ngừa và điều trị bằng cách kiểm soát huyết áp sau mổ và không dùng đường uống ở giai đoạn sớm sau phẫu thuật
Thông tim can thiệp
Trong gần 40 năm, phẫu thuật là lựa chọn điều trị duy nhất cho bệnh nhân hẹp eo động mạch chủ. Vào những năm đầu thập niên 80, tạo hình mạch máu bằng bóng qua da đă được thực hiện lần đầu tiên thành công ở sơ sinh [37]. Gần đây, can thiệp thông tim với tạo hình mạch máu bằng bóng ( có hoặc không có đặt stent ) được thực hiện ngày càng nhiều và có kết quả tốt đối với hẹp eo động mạch chủ nguyên gốc và tái phát.(h́ình 8.5a,b)
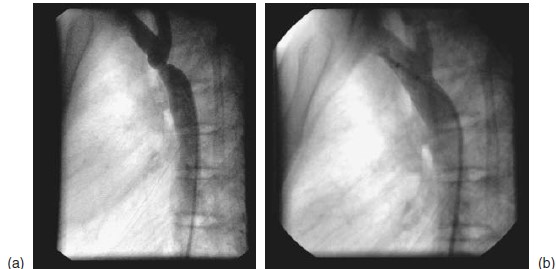
Hình 8.5: Hình ảnh thông tim của hẹp eo động mạch chủ nguyên gốc trước đặt stent (a) và sau khi đặt stent , (b)ghi nhận chỗ hẹp eo đã được giải phóng.
Ban đầu, tạo hình mạch máu bằng bóng có tỷ lệ thành công ngắn hạn khá tốt đối với hẹp eo động mạch chủ nguyên gốc, nhưng có tỷ lệ tái hẹp và tạo phình tương đối cao khi so sánh với phẫu thuật. Kỹ thuật này cũng làm tăng nguy cơ bóc tách và vỡ động mạch chủ [38-41]. Trong các nghiên cứu theo dõi trung hạn, ý nghĩa lâm sàng của các túi phình nhỏ chưa được xác định vì phần lớn không cần can thiệp. Tuy nhiên sự tồn tại của mô động mạch chủ bất thường, các mô này sẽ được lấy đi khi phẫu thuật, có thể làm tăng nguy cơ tạo phình hoặc bóc tách.
Đặt stent nội mạc khi nong động mạch chủ được cho là cải thiện kết quả can thiệp vì có một cấu trúc nâng đỡ cho mạch máu được nong, giảm sự dãn nở quá mức của động mạch chủ và pḥòng ngừa co dội ngược động mạch chủ [38].
Ở trẻ em và người lớn, các nghiên cứu ngắn hạn cho thấy nong và đặt stent đối với hẹp eo động mạch chủ nguyên gốc và tái phát có tỷ lệ giải phóng chỗ hẹp thành công lên đến 98%. Tuy nhiên, kỹ thuật này cũng có tỷ lệ biến chứng cấp khá cao (14%). Mặc dù phần lớn các biến chứng nhẹ và không cần can thiệp. Biến chứng bao gồm tạo phình động mạch chủ, bóc tách động mạch chủ và tử vong [42]. Các nghiên cứu khác ghi nhận kết quả tương tự trong giải phóng tắc nghẽn và tăng huyết áp ở bệnh nhân phẫu thuật, nhưng bệnh nhân đặt stent có tỷ lệ tái phát và cần can thiệp lại cao hơn [43]. Một số bệnh nhân có xơ vữa động mạch, viêm mạch máu, bệnh mô liên kết cần phải được cân nhắc khi quyết định can thiệp vì có cấu trúc mô động mạch chủ bất thường, những bệnh nhân này có nguy cơ biến chứng cao. Tóm lại, kết quả nong và đặt stent đang được quan tâm và có nhiều triển vọng, giúp tránh vết mổ và thời gian hồi phục lâu sau mổ, chỉ định can thiệp thông tim trên hẹp eo động mạch chủ nguyên gốc vẫn c̣òn bàn căi.
Hẹp eo động mạch chủ tái phát
Tái hẹp eo động mạch chủ tương đối thường gặp, ít nhất 5-10% tổng số bệnh nhân [23-28]. Tỷ lệ cao nhất ở bệnh nhân mổ sơ sinh, lên đến 44%. Sự tái hẹp liên quan nhiều nhất đến sự tạo thành sẹo và mô sợi tại chỗ được sửa chữa. Kỹ thuật mới cắt bỏ đoạn hẹp kèm nối tận-tận có mở rộng có thể giúp giảm tỷ lệ tái hẹp.
Khi nghi ngờ tái hẹp, đánh giá và chỉ định can thiệp cũng tương tự như trong hẹp eo động mạch chủ nguyên gốc. Tuy nhiên, ở bệnh nhân sau mổ, siêu âm có thể khó khăn và nên tìm thêm các di chứng sau can thiệp, chẳng hạn như sự tạo phình.
Tùy theo bệnh cảnh lâm sàng và giải phẫu học, tái hẹp eo có thể được điều trị bằng phẫu thuật hoặc thông tim. Trong một vài trường hợp như có phình động mạch chủ, vị trí tái hẹp gần chỗ xuất phát động mạch cảnh hoặc có các tổn thương phối hợp như hẹp van động mạch chủ, phẫu thuật là lựa chọn thích hợp hơn [44]. Khi bệnh nhân có kèm theo dãn động mạch chủ, bệnh van động mạch chủ hoặc bệnh mạch vành, phẫu thuật có thể được thực hiện theo một đường mổ giữa xương ức, tạo cầu nối giữa động mạch chủ lên và động mạch chủ xuống [45].
Tuy nhiên, hiện nay phần lớn trường hợp tái hẹp eo động mạch chủ sau mổ được điều trị bằng thông tim can thiệp, nong chỗ hẹp bằng bóng kèm hoặc không kèm đặt stent. Đối với tái hẹp eo, thông tim can thiệp có tỷ lệ thành công 80%. Trong trường hợp này, tỷ lệ phình, rách và vỡ động mạch chủ thấp hơn hẹp eo động mạch chủ nguyên gốc ( ~1.5%) [38]. Tần suất biến chứng giảm được cho là do các mô bất thường đã được lấy đi ở phẫu thuật trước đó và sự tạo sẹo trên thành động mạch chủ. Mặc dù thông tim được lựa chọn nhiều trong can thiệp tái hẹp eo, nhưng vẫn chưa có nghiên cứu lớn ngẫu nhiên, có đối chứng so sánh giữa phẫu thuật và thông tim can thiệp trong nhóm dân số này.
Tóm tắt về can thiệp
Quyết định lựa chọn phương pháp can thiệp trên hẹp eo động mạch chủ nguyên gốc và tái phát không hề dễ dàng, đòi hỏi phải cân nhắc giữa lợi ích và nguy cơ của từng phương pháp can thiệp trên mỗi bệnh nhân. Vấn đề quan trọng khi lựa chọn là kinh nghiệm trong từng phương pháp can thiệp, biến chứng sau thủ thuật và kết quả lâu dài. Phần lớn trẻ sơ sinh và trẻ em dưới 1 tuổi sẽ được phẫu thuật đối với hẹp eo động mạch chủ nguyên gốc. Đối với hẹp eo động mạch chủ tái phát, có sự đồng thuận chung cho rằng nên nong bằng bóng kèm hoặc không kèm đặt stent nếu cấu trúc giải phẫu thuận lợi. Ở trẻ lớn và người lớn, cả 2 phương pháp thông tim can thiệp và phẫu thuật đều có thể được lựa chọn. Cuối cùng, lựa chọn phương pháp can thiệp sẽ phụ thuộc vào cấu trúc giải phẫu và nguyện vọng của từng bệnh nhân, cân nhắc các nguy cơ cũng như là kinh nghiệm của phẫu thuật viên và bác sĩ thông tim can thiệp. Nếu can thiệp bằng thông tim được lựa chọn thì thủ thuật chỉ nên được thực hiện ở trung tâm có nhiều kinh nghiệm và có hỗ trợ phẫu thuật tim bẩm sinh.
Các vấn đề nội khoa phối hợp
Tăng huyết áp
Bệnh nhân hẹp eo động mạch chủ chưa sửa chữa thường có tăng huyết áp. Tăng huyết áp có thể vẫn tồn tại sau khi đã giải phóng chênh áp qua chỗ hẹp eo, nhất là khi sửa chữa trễ ở trẻ lớn và người lớn [23,25-27,46]. Tỷ lệ tăng huyết áp cao trong trường hợp không có hẹp eo động mạch chủ tồn lưu có thể do bất thường mạch máu làm tổn thương phản ứng mạch máu ngoại biên và gia tăng độ cứng động mạch chủ [46,48]. Một yếu tố khác có thể góp phần làm tăng huyết áp là tình trạng tăng động thất trái, nhất là khi thất trái bị phì đại.
Đánh giá huyết áp tại mỗi lần thăm khám là rất quan trọng ở bệnh nhân có hẹp eo động mạch chủ. Bên cạnh đo huyết áp thường quy, một số bác sĩ c̣òn đo huyết áp di động 24 giờ để chẩn đoán tăng huyết áp trung bình [49]. Tăng huyết áp làm tăng nguy cơ tử vong sớm do làm tăng nguy cơ bệnh mạch vành sớm và tai biến mạch máu năo. Vì vậy, tăng huyết áp phải được điều trị ngay khi chẩn đoán.
Ngoài tăng huyết áp lúc nghỉ, vài bệnh nhân còn có tăng huyết áp khi gắng sức. Ý nghĩa của đáp ứng huyết áp này và nhu cầu điều trị vẫn còn bàn căi. Mối liên quan giữa huyết áp đỉnh khi gắng sức và hậu quả lâu dài hoặc huyết áp di động 24 giờ chưa được xác định [50-53]. Nhiều bác sĩ chuyên khoa bệnh tim bẩm sinh người lớn điều trị tăng huyết áp khi gắng sức bằng thuốc, thường là ức chế bêta, nhằm mục đích làm giảm áp lực tác động lên động mạch chủ, áp lực này có thể ảnh hưởng lên đoạn động mạch chủ đã sửa chữa và có thể gây phình, bóc tách hoặc vỡ.
Bệnh động mạch vành
Bệnh động mạch vành sớm là nguyên nhân tử vong thường gặp ở bệnh nhân hẹp eo động mạch chủ [23,25-27]. Cơ chế bệnh sinh của hiện tượng này chưa được giải thích đầy đủ, có thể liên quan một phần đến tăng huyết áp. Một số cơ chế nội tại động mạch cũng có thể ảnh hưởng đến cơ chế sinh bệnh. Chúng ta biết rằng những bệnh nhân có tổn thương phản ứng vận mạch thì có nguy cơ bệnh mạch vành, vài nghiên cứu cho thấy một số bệnh nhân hẹp eo có bất thường chức năng mạch máu này [14,47,54]. Những bệnh nhân hẹp eo động mạch chủ có hoặc không có sửa chữa đều nên tầm soát các yếu tố nguy cơ bệnh mạch vành quan trọng và thay đổi lối sống, bao gồm kiểm tra mức độ cholesterol, ngưng thuốc lá, tầm soát tiểu đuờng. Bất kỳ triệu chứng đau ngực nào cũng nên đuợc đánh giá đầy đủ tổn thương động mạch vành.
Phình mạch máu não
Một biến chứng khác và là nguyên nhân tử vong ở bệnh nhân hẹp eo động mạch chủ là xuất huyết duới màng nhện do phình mạch máu. Vào đầu những năm 1900, bác sĩ Maude Abbott ghi nhận đây là một nguyên nhân tử vong thường gặp trong các nghiên cứu tử thiết [22]. Gần đây, một loạt bệnh nhân có sửa chữa hẹp eo động mạch chủ được tâm soát phình mạch máu não tại bệnh viện Mayo [55], kết quả cho thấy trong 100 bệnh nhân thì 10 bệnh nhân có phình , trong đó 9 bệnh nhân không có triệu chứng. Phần lớn tổn thương phình nhỏ nhưng xuất độ thì cao hơn dân số chung. Tỷ lệ vỡ của các tổn thương phình mạch máu nhỏ thấp, ít nhất là trong thời gian ngắn. Tuy nhiên, vấn đề điều trị cho từng bệnh nhân cần hội chẩn với bác sĩ chuyên khoa trong lĩnh vực này. Hiện nay chưa có nghiên cứu về phình mạch máu ở trẻ em hoặc bệnh nhân người lớn đã được sửa chữa sớm. Cho đến thời điểm nay, tính ứng dụng và lợi ích của tầm soát thường quy phình mạch máu não ở bệnh nhân hẹp eo động mạch chủ vẫn chưa rõ ràng. Đây vẫn còn là một vấn đề được quan tâm trong tương lai.
Phình và bóc tách động mạch chủ
Phình và bóc tách động mạch chủ có thể xảy ra ở vùng sửa chữa hẹp eo động mạch chủ trước đó hoặc có liên quan đến các thủ thuật thông tim. Có sự kết hợp giữa phình và bóc tách động mạch chủ lên với hẹp eo động mạch chủ. Phần lớn xảy ra ở bệnh nhân có van động mạch chủ 2 mảnh. Hiện nay chưa rõ bệnh nhân có hẹp eo động mạch chủ kèm van động mạch chủ 2 mảnh có nguy cơ cao hơn bệnh nhân có van động mạch chủ 2 mảnh đơn thuần hay không. Phình và bóc tách động mạch chủ lên cũng được mô tả ở bệnh nhân hẹp eo động mạch chủ kèm tăng huyết áp không được kiểm soát tốt [24,56].
Theo dõi thường quy
Rõ ràng hẹp eo động mạch chủ là tổn thương lâu dài và không chỉ ảnh hưởng lên đoạn động mạch chủ hẹp, bệnh nhân cần được theo dõi sát sau can thiệp bằng phẫu thuật hoặc thông tim ( bảng 8.1). Bệnh nhân được khuyến cáo nên theo dõi tại trung tâm chuyên khoa về bệnh tim bẩm sinh người lớn [57]. Bệnh nhân được theo dõi ít nhất mỗi năm, bao gồm hỏi bệnh sử chi tiết để loại trừ đau ngực, suy tim và đau cách hồi, bắt mạch tứ chi và đo huyết áp. Siêu âm được thực hiện để tầm soát tái hẹp eo, phình động mạch chủ , đánh giá chức năng và sự phì đại thất trái.
Bảng 8.1: Đánh giá theo dõi thường quy đối với bệnh nhân hẹp eo động mạch chủ
|
Bệnh sử |
Đánh giá mỗi năm các triệu chứng: đau ngực, không dung nạp với gắng sức, suy tim, đau cách hồi |
|
Khám lâm sàng |
Đánh giá mỗi năm: âm thổi mới, mạch tứ chi, huyết áp |
|
Siêu âm |
Đánh giá mỗi năm: động mạch chủ lên, van động mạch chủ, kích thước/ độ dày thành/ chức năng thất trái, chênh áp qua van động mạch chủ, doppler động mạch chủ xuống và động mạch chủ bụng. |
|
MRI và CT scan |
Tần suất thực hiện thay đổi, thường xuyên hơn với bênh nhân có nguy cơ phình cao. |
|
Nghiệm pháp gắng sức |
Tần suất thực hiện thay đổi, dành cho bệnh nhân có tăng huyết áp. |
|
Theo dõi huyết áp di động 24 giờ |
Tần suất thực hiện thay đổi, dành cho bệnh nhân có tăng huyết áp. |
|
Thông tim |
Thường thực hiện khi cần can thiệp |
CT: Computed tomography (chụp cách lớp điện toán).
MRI: Magnetic resonance imaging (ảnh cộng hưởng từ).
Các chẩn đoán hình ảnh khác như CT scan và MRI, được thực hiện phụ thuộc từng tình huống lâm sàng, mặc dù khi chụp CT scan thì bệnh nhân phải tiếp xúc tia xạ đáng kể. Bệnh nhân đã sửa chữa hoặc can thiệp, cần thực hiện ít nhất một xét nghiệm chẩn đoán hình ảnh để tầm soát sự tạo phình . Tần xuất thực hiện lại các cận lâm sàng này còn thay đổi và chưa có tiêu chuẩn dựa trên chứng cứ nào. Đối với bệnh nhân có nguy cơ cao bị phình động mạch (ví dụ: tạo hình động mạch chủ bằng miếng vá, phẫu thuật ở tuổi cao, nong bằng bóng) thì nên thực hiện thường xuyên hơn. Đối với bệnh nhân đã đặt stent, nên làm CT hơn MRI vì stent gây nhiễu hình ảnh MRI.
Đo huyết áp rất quan trọng tại mỗi lần thăm khám. Như đã đề cập ở trên, nên làm trắc nghiệm gắng sức hoặc đo huyết áp 24 giờ để tầm soát. Bất cứ bằng chứng tăng huyết áp nào đều nên điều trị để đạt mức độ huyết áp bình thường..
Nhiều bệnh nhân, đặc biệt là bệnh nhân đang học trung học và đại học, quan tâm nhiều đến khả năng hoạt động thể lực. Các bệnh nhân này được khuyến khích tập thể dục và vận động thể lực, một phần vì họ có nguy cơ cao tăng huyết áp và bệnh động mạch vành sớm. Tuy nhiên, cần giới hạn các môn thể thao có đối kháng ngay cả khi bệnh nhân đã được sửa chữa, tùy thuộc vào tổn thương tồn lưu và các tổn thương phối hợp. Các khuyến cáo về việc tham gia các môn thể thao đối kháng đã được xem xét lại trong hướng dẫn đồng thuận Bethesda lần thứ 36 của trường môn tim mạch Hoa Kỳ [58].
Khi bệnh nhân đã sửa chữa cần can thiệp nội khoa hoặc ngoại khoa, thường có phối hợp nhiều tình trạng như tăng huyết áp, hẹp eo động mạch chủ tái phát, bệnh động mạch vành, phình mạch máu hoặc bệnh van tim. Tiếp cận kỹ lưỡng theo từng bệnh nhân là bắt buộc. Điều này giúp theo dõi đều đặn, can thiệp sớm và thay đổi các yếu tố nguy cơ. Nhờ đó mà tỷ lệ sống sót và chất lượng cuộc sống tiếp tục được cải thiện
BÊNH ĐỘNG MẠCH CHỦ
Định nghĩa và bệnh học
Bệnh động mạch chủ được định nghĩa là tất cả các bệnh lý của động mạch chủ. Chương này tập trung vào các bệnh lý di truyền của lớp trung mạc động mạch chủ ( bảng 8.2), bệnh van động mạch chủ 2 mảnh và sự phát triển kết hợp của phình động mạch chủ. Phình động mạch chủ được xác định khi kích thước động mạch chủ > 1.5 bình thường và bệnh nhân có nguy cơ bị bóc tách và vỡ động mạch chủ, đe dọa tính mạng. Chương này tập trung mô tả các bệnh lý di truyền này và đưa ra cách tiếp cận chung để tầm soát, theo dõi và can thiệp.
Các bệnh lý di truyền liên quan đến bất thường mô liên kết động mạch chủ thường gặp nhất. Đây là sự thoái hóa và mất cân bằng giữa cơ trơn, collagen, elastin và fibrillin trong trung mạc động mạch chủ. Dấu hiệu mô học “ hoại tử trung mô dạng nang” đã được tìm thấy trong các bệnh lý về gen và cũng được thấy trong động mạch chủ ở bệnh nhân có van động mạch chủ 2 mảnh, lớn tuổi và tăng huyết áp [59].
Bảng 8.2: Bệnh mô liên kết di truyền có liên quan đến động mạch chủ
|
Hội chứng Marfan Bệnh ly fibrillin Hội chứng Ehlers-Danlos ( type I, II, và IV) Đột biến thay đổi thụ thể yếu tố tăng trưởng bêta (Hội chứng Loeys-Dietz) Phình và bóc tách động mạch chủ ngực có tính gia đình Hội chứng Turner |
Bệnh mô liên kết
Hội chứng Marfan
Hội chứng Marfan được mô tả là rối loạn mô liên kết di truyền trội trên nhiễm sắc thể thường, gây ra bởi sự đột biến gen fibrillin 1, biểu hiện ở nhiều phenotype khác nhau, Biểu hiện lâm sàng chủ yếu là hệ tim mạch, xương và mắt. Chẩn đoán dựa vào biểu hiện lâm sàng, sử dụng phân loại bệnh học có sửa đổi của Ghent [60]. Trắc nghiệm gen về sự đột biến fibrillin cũng có giá trị.
Biểu hiện tim mạch chính của hội chứng Marfan là dãn gốc động mạch chủ ( hình 8.6a) và sa van 2 lá. Dãn động mạch chủ thường xảy ra tại xoang động mạch chủ (xoang Valsalva), động mạch chủ có nguy cơ bị bóc tách và vỡ, bệnh nhân cũng có thể bị phình và bóc tách động mạch chủ xuống nhưng ít gặp hơn [61].
Một số hội chứng khác có liên quan đến liên quan đến bất thường fibrillin và có thể có dãn động mạch chủ nhưng không có các tiêu chuẩn chẩn đoán lâm sàng của hội chứng Marfan. Bao gồm kiểu hình (phenotype) MASS ( cận thị, dãn động mạch chủ, dấu hiệu da hoặc xương không đặc hiệu), hội chứng phình động mạch chủ có tính gia đình, hội chứng sa van 2 lá [62].
Hội chứng Ehlers-Danlos
Hội chứng Ehlers-Danlos có nhiều type với nhiều biểu hiện lâm sàng khác nhau. Type thường kết hợp với biểu hiện động mạch chủ nặng là type IV hoặc còn gọi là type ác tính. Đây là rối loạn trội trên nhiễm sắc thể thường với bất thường type collagen III. Bệnh nhân hội chứng Ehlers-Danlos type IV có nguy cơ bị bóc tách các mạch máu lớn, bao gồm động mạch chủ, động mạch não và động mạch bụng, không có các biểu hiện da và khớp của dạng kinh điển thường gặp [63].
Dạng hội chứng Ehlers-danlos kinh điển, tức type I và II, có biểu hiện da mỏng và dãn qúa mức, một số ít có kèm sa van 2 lá và dãn động mạch chủ. Những bệnh nhân này ít có nguy cơ phình và bóc tách động mạch chủ hơn type IV. Dạng thường gặp nhất của hội chứng này là type III, type này ít có biểu hiện tại động mạch chủ và không cần theo dõi tim mạch.
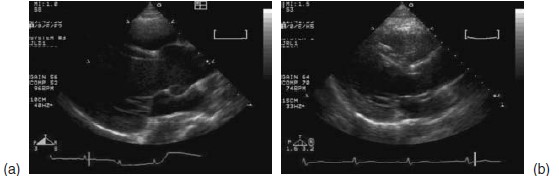
Hình 8.6: Hình ảnh dãn động mạch chủ trên siêu âm
a) xoang vành dãn ở bệnh nhân hội chứng Marfan
b) Mờ chỗ nối xoang ống và dãn đoạn gần động mạch chủ lên ở bệnh nhân van động mạch chủ 2 mảnh
Đột biến thay đổi thụ thể yếu tố tăng trưởng bêta
(Hội chứng Loeys-Dietz)
Đột biến thay đổi thụ thể yếu tố tăng trưởng bêta được coi là những tình trạng có kèm dãn và bóc tách động mạch chủ. Gần đây, đột biến thay đổi thụ thể yếu tố tăng trưởng bêta loại 1 và 2 được mô tả là hội chứng Loeys-Dietz. Hội chứng này có biểu hiện đặc hiệu trên tim, xương và thần kinh, bao gồm tăng khoảng cách 2 mắt, lưỡi gà chẻ đôi, xoắn động mạch và phình động mạch chủ. Hội chứng Loeys-Dietz là loại bệnh động mạch chủ tiến triển khá nặng với tỷ lệ tạo phình và tử vong sớm cao. Trong một nghiên cứu hồi cứu, 6 trong 71 bệnh nhân bị bóc tách và vỡ động mạch chủ khi đường kính động mạch chủ <4.5 cm và xảy ra khi mới 6 tuổi [64,65].
Hội chứng phình và bóc tách động mạch chủ ngực.
Hội chứng phình và bóc tách động mạch chủ ngực có tính gia đình là một rối loạn về gen, di tryền trội trên nhiễm sắc thể thường kèm kiểu hình thay đổi. Trong hội chứng này, có sự tập hợp bệnh động mạch chủ có tính gia đình mà không kèm theo biểu hiện lâm sàng của các hội chứng khác. Một nghiên cứu gần đây cho thấy, bệnh có thể kết hợp với đột biến thay đổi thụ thể yếu tố tăng trưởng bêta [66].
Hội chứng Turner
Hội chứng Turner là một rối loạn về gen (45,X) phối hợp với tăng huyết áp, hẹp eo động mạch chủ và van động mạch chủ 2 mảnh. Dãn gốc động mạch chủ xảy ra trong 40% trường hợp và có nguy cơ bóc tách động mạch chủ [9,10,67,68]. Nguy cơ này được cho là do có tồn tại các bất thường tim mạch khác đi kèm. Động mạch chủ giảm độ đàn hồi và bất thường mô nội tại có liên quan đến bóc tách động mạch chủ. Tuy nhiên, không giống các bệnh ly khác, bất thường này có thể liên quan đến các bất thường hệ bạch huyết trong hội chứng Turner [69].
Van động mạch chủ 2 mảnh
Van động mạch chủ 2 mảnh là một bệnh lý thường gặp, chiếm 1-2% dân số chung. Bên cạnh bất thường van, bệnh nhân còn tăng nguy cơ phình gốc động mạch chủ và động mạch chủ lên [70]. Khác với hội chứng Marfan với phần lớn bệnh nhân phình tại xoang valsalva, bệnh nhân có van động mạch chủ 2 mảnh thường bị dãn tại chỗ nối xoang ống và đoạn gần động mạch chủ lên ( hình 9.6b). Bệnh nhân có biểu hiện dãn động mạch chủ ngay cả khi huyết động qua van động mạch chủ bình thường. Tuy nhiên, khi có tổn thương khác làm thay đổi huyết động như hẹp hoặc hở van động mạch chủ thì khả năng dãn và phình động mạch chủ có khuynh hướng tăng [70,71].
Mặc dù không phải là rối loạn mô liên kết điển hình nhưng bệnh van động mạch chủ 2 mảnh cũng được đề cập trong chương này vì những nghiên cứu mô học gần đây cho thấy có bất thường mô nội tại trong động mạch chủ lên. Sự mất điều hòa lớp trung mạc được xác định bởi bất thường tế bào cơ trơn, mỏng lớp elastic, bất thường đáp ứng viêm hoặc có biểu hiện metalloproteinase trong chất căn bản (matrix). So với van động mạch chủ 3 mảnh, mô động mạch chủ của van động mạch chủ 2 mảnh có thay đổi và bất thường chức năng lớp elastic. Điều này làm giảm hoạt tính vận mạch và giảm dãn năng [70-73].
Tầm soát và theo dõi
Bệnh động mạch chủ khó chẩn đoán vì triệu chứng không điển hình cho đến khi bệnh nhân có các biến chứng nặng như bóc tách hoặc vỡ. Bệnh nhân được chẩn đoán sớm thường nhờ vào tiền căn gia đình có bệnh động mạch chủ hoặc có rối loạn về gen, hoặc do biểu hiện của một tình trạng khác có liên quan. Khi bệnh nhân có nguy cơ bệnh động mạch chủ, cần thăm khám định kỳ để phát hiện sự hình thành và phát triển của phình động mạch chủ. Đối với tất cả các tình trạng được đề cập ở trên, vấn đề tầm soát được khuyến cáo giống nhau trừ khi có bất thường khác đi kèm. Đối với bệnh nhân có van van động mạch chủ 2 mảnh, nên tầm soát những người thân thế hệ thứ nhất vì gần 20% trong số này có van động mạch chủ 2 mảnh hoặc bất thường tim mạch khác.
Siêu âm tim qua thành ngực được khuyến cáo nên làm mỗi năm. Những bệnh nhân có bệnh động mạch chủ nặng, tổn thương van đi kèm hoặc kích thước động mạch chủ tiến triển nhanh ( >0.5 cm/ 6 tháng) nên được đánh giá ít nhất mỗi 6 tháng. Phần lớn bệnh nhân nên chụp MRI hoặc CT scan ngay từ đầu và việc thực hiện lại thay đổi tùy theo từng bệnh nhân. Nên lập lại thường hơn nếu bệnh nhân có thay đổi đáng kể trên siêu âm qua thành ngực, có triệu chứng hoặc cửa sổ siêu âm kém. Vấn đề tiếp xúc tia xạ khi chụp CT cần được quan tâm vì bệnh nhân thường trẻ và vài bệnh nhân phải chụp thường mỗi 1-2 năm [75]. Siêu âm qua thực quản cũng là một phương pháp đánh giá động mạch chủ, tuy nhiên, phương pháp này xâm nhập hơn CT và MRI và không cho biết thêm nhiều thông tin. Siêu âm qua thực quản thường được dùng đánh giá bóc tách động mạch chủ trong tình huống cấp cứu.
Điều trị
Điều trị nội khoa hoặc ngoại khoa phình động mạch chủ trong các rối loạn mô liên kết thay đổi tùy theo bệnh căn bản. Bên cạnh một số bệnh lý có khuyến cáo rõ ràng như hội chứng Marfan, các rối loạn khác vẫn chưa có sự thống nhất, chẳng hạn như bệnh van động chủ 2 mảnh. Phần lớn các rối loạn di truyền không thường gặp nên nhiều khuyến cáo được rút ra từ tài liệu về hội chứng Marfan và bóc tách động mạch chủ trong dân số chung.
Điều trị nội khoa
Điều trị nội khoa căn bản của dãn động mạch chủ trong hội chứng Marfan là ức chế bêta, điều trị tăng huyết áp tích cực và thay đổi lối sống, Thuốc ức chế bêta được dùng nhằm làm chậm tiến triển của dãn động mạch chủ và giảm áp lực xé lên thành động mạch. Trong những năm gần đây, hiệu quả của ức chế bêta đang là một vấn đề và có thông tin cho thấy thuốc ức chế men chuyển có lợi trong vấn đề làm chậm tiến triển gốc động mạch chủ [76,77]. Mới hơn nữa là một thử nghiệm trên chuột cho thấy hiệu quả của thuốc ức chế thụ thể angiotensine, các thử nghiệm này vẫn đang được tiến hành [78,79].
Những can thiệp nội khoa không xâm nhập bao gồm giáo dục gia đình, tham vấn về những thận trọng đối với các môn thể thao đối kháng và vận động thể lực, chẳng hạn như giới hạn các hoạt động va chạm mạnh hoặc thực hiện các hoạt động tĩnh tại [80,81]. Cần giáo dục bệnh nhân và người nhà những triệu chứng quan trọng như đau ngực hoặc đau lưng vì đó là triệu chứng bóc tách động mạch chủ. Không nên có thai nếu đường kính gốc động mạch chủ trên 4 cm.
Đối với các rối loạn mô liên kết khác thì chưa có khuyến cáo dựa trên chứng cứ. Điều trị tăng huyết áp là quan trọng cho tất cả bệnh nhân bệnh động mạch chủ. Tuy nhiên, chưa có khuyến cáo toàn cầu về việc sử dụng ức chế beta trong các bệnh mô liên kết ngoại trừ hội chứng Marfan. Thuốc ức chế thụ thể angiotensin có thể có lợi trong hội chứng Loeys-Dietz và một số tác giả khuyến cáo nên sử dụng thuốc này. Vấn đề hạn chế vận động và các triệu chứng cảnh báo quan trọng trong hội chứng Marfan cũng tương tự như trong các rối loạn khác. Có nhiều khuyến cáo khác nhau về vận động đối với bệnh nhân van động mạch chủ 2 mảnh và dãn động mạch chủ [82]
Điều trị ngoại khoa
Bệnh nhân hội chứng Marfan có nguy cơ bóc tách động mạch chủ khi đường kính động mạch chủ đạt một kích thước nhất định hoặc tiến triển nhanh, do đó phẫu thuật dự phòng có thể được đặt ra. Những tiêu chuẩn phẫu thuật được rút ra từ các nghiên cứu hồi cứu về bóc tách động mạch chủ và hội chứng Marfan. Khi đường kính động mạch chủ trên 6 cm thì có 10% nguy cơ bóc tách hoặc vỡ trong năm tiếp theo, bất kể bệnh nguyên của dãn động mạch chủ [83,84]. Do đó, phẫu thuật dự phòng thường được khuyến cáo khi đường kính động mạch chủ dãn 5 cm hoặc tiến triển nhanh ( >0.5 cm/6 tháng) [61]. Hội tim mạch Châu Âu khuyến cáo phẫu thuật khi đường kính động mạch chủ trong hội chứng Marfan 4.5 cm [85]. Nếu có hở van động mạch chủ trung bình tới nặng hoặc có tiền căn gia đình bị bóc tách động mạch chủ ở kích thước nhỏ hơn, thì nên xem xét phẫu thuật sớm hơn.
Không có nhiều tài liệu về các bệnh mô liên kết, tuy nhiên, dựa vào dữ liệu về bóc tách động mạch chủ trong dân số chung, tiêu chuẩn 5 cm được dùng để xem xét phẫu thuật dự phòng. Những khuyến cáo về phẫu thuật van động mạch chủ 2 mảnh có dãn động mạch chủ đã được bàn luận trong các hướng dẫn mới nhất của Hội Tim Mạch Hoa Kỳ. Theo hướng dẫn này, phẫu thuật khi đường kính động mạch chủ 5 cm hoặc tốc độ tiến triển trên 0.5 cm/năm. Thay gốc động mạch chủ có thể thực hiện ở đường kính nhỏ hơn nếu cần phẫu thuật van động mạch chủ [86].
Vài trường hợp cho phép phẫu thuật khi đường kính động mạch chủ nhỏ hơn 5 cm. Vài báo cáo cho thấy hội chứng Loeys-Dietz có biểu hiện bệnh động mạch chủ nặng và phẫu thuật nên thực hiện khi đường kính động mạch chủ ở người lớn là 4 cm [64,65]. Trong hội chứng Turner, bệnh nhân nữ thường có thể tạng thấp hơn dân số chung và bóc tách được ghi nhận ở đường kính < 5cm. Trong trường hợp này, dùng chỉ số đo kích thước động mạch chủ để xem xét phẫu thuật, ví dụ > 2.5 cm/m2 diện tích cơ thể [10].
Khi thực hiện phẫu thuật dự phòng, điều quan trọng là phải có phẫu thuật viên lồng ngực có kinh nghiệm về bệnh mô liên kết. Các phẫu thuật này có thể khó hơn do có bất thường động mạch chủ kết hợp. Thay gốc động mạch chủ có thể thực hiện kèm hoặc không kèm thay van động mạch chủ. Nếu cần thay van như trong trường hợp hội chứng Marfan kèm hở van động mạch chủ hoặc van động mạch chủ 2 mảnh kèm hẹp hay hở van động mạch chủ, thường thực hiện phẫu thuật Bentall. Phẫu thuật này kết hơp thay gốc động mạch chủ và động mạch chủ lên từ van cho tới thân cánh tay đầu kèm cắm lại động mạch vành [87]. Một van nhân tạo được đặt bên trong mảnh ghép và bệnh nhân phải uống thuốc kháng đông suốt đời. Phẫu thuật này có tỷ lệ sống sót sau 5, 10 và 20 năm lần lượt là 88%, 81% và 75% [88]. Gần đây, thủ thuật giữ lại van đã được thực hiện, trong đó, động mạch chủ được thay từ đoạn xoang đến động mạch thân cánh tay đầu bằng một ống ghép và cắm lại động mạch vành. Van động mạch chủ nguyên vẹn được cấy vào ống ghép hoặc để vị trí cũ dưới ống ghép [89-91].Thời gian phẫu thuật và nguy cơ liên quan cần được bàn luận kỹ giữa phẫu thuật viên, bác sĩ tim mạch và bệnh nhân để đưa ra quyết định tốt nhất cho từng bệnh nhân.
Tóm tắt trường hợp bệnh
Trường hợp bênh nhân được nêu ở đầu chương này là một bệnh nhân nữ 23 tuổi có hẹp tái phát và phình động mạch chủ tại vị trí sửa chữa hẹp eo trước đó. Bệnh nhân có chênh áp tồn lưu đáng kể qua huyết áp và siêu âm. Lựa chọn can thiệp cho bệnh nhân này thì phức tạp vì có nhiều yếu tố, bao gồm 2 lần phẫu thuật , có phình và vị trí của các mạch máu đầu và cổ liên quan đến chỗ hẹp. Bệnh nhân cũng có bệnh sử chậm phát triển và có tổn thương não, có thể liên quan đến các phẫu thuật trước đó. Vì những lý do đó, một phương pháp điều trị phối hợp đã được lựa chọn. Bệnh nhân được can thiệp phẫu thuật, đầu tiên là đặt lại động mạch thân cánh tay đầu và động mạch cảnh chung trái về đoạn gần của động mạch chủ, sau đó bệnh nhân được đặt stent nội mạch ngay trong lúc phẫu thuật để giải phóng chỗ hẹp eo. Phẫu thuật được thực hiện không dùng tuần hoàn ngoài cơ thể và duy trì tưới máu não. Bệnh nhân đã có kết quả tốt với chênh áp < 5 mmHg trong phòng mổ , chênh áp trung bình 12 mmHg qua siêu âm và không có phổ tâm trương, chênh lệch huyết áp 14 mmHg. CT scan theo dõi sau 3 tháng cho thấy stent rõ, không có hẹp eo động mạch chủ tồn lưu và không có phình mới xuất hiện.
Tài liệu tham khảo
- Sadler TW. Langman’s Medical Embryology (6th edition). Baltimore: Williams and Wilkins, 1990.
- Knight L, Edwards J. Right aortic arch: types and associated cardiac anomalies. Circulation 1974;50:1047–51.
- KumarV, Fausto N, Abbas A. Robbins andCotran Pathologic Basis of Disease (7th edition). Philadelphia: Elsevier, 2005.
- Aboulhosn J, Child JS. Left ventricular outflow obstruction, subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation 2006;114:2412–22.
- Keith JD. Coarctation of the aorta. In: Keith JD, Rowe RD, Vlad P, eds. Heart Disease in Infancy and Childhood. New York: Macmillan, 1978:736–57.
- Fyler DC, Buckley LP, Hellenbrand WE, et al. Report of the New England regional infant cardiac program. Pediatrics 1980;65:432–6.
- McBride KL, Pignatelli R, Lewin M, et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am J Med Genet A 2005;134(2):180–6.
- Wessels MW, Berger RM, Frohn-Mulder IM, et al. Autosomal dominant inheritance of left ventricular outflow tract obstruction. Am J Med Genet A 2005;134(2):171–9.
- Gotzsche CO, Krag-Olsen B, Nielsen J, et al. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child 1994; 71(5):433–6.
- Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation 2007;116:1663–70.
- Nihoyannopoulos P, Karas S, Sapsford RN, et al. Accuracy of two-dimensional echocardiography in the diagnosis of aortic arch obstruction. J Am Coll Cardiol 1987; 10(5):1072–7.
- Fyler D. Coarctation of the aorta. In: Nadas’ Pediatric Cardiology. Philadelphia: Hanley and Belfus, 1992:535–55.
- Rudolph AM, Heymann MA, Spitznas U. Hemodynamic considerations in the development of narrowing of the aorta. Am J Cardiol 1972;30:514.
- Vogt M, Kuhn A, Baumgartner D, et al. Impaired elastic properties of the ascending aorta in newborns before and early after successful coarctation repair: proof of a systemic vascular disease of the prestenotic arteries? Circulation 2005;111(24):3269–73.
- Niwa K, Perloff JK, Bhuta SM, et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation 2001; 103(3):393–400.
- Isner JM, Donaldson RF, Fulton D, et al. Cystic medial necrosis in coarctation of the aorta: a potential factor contributing to adverse consequences observed after percutaneous balloon angioplasty of coarctation sites. Circulation 1987;75(4):689–95.
- Nicol ED, Gatzoulis M, Padley SP, Rubens M. Assessment of adult congenital heart disease with multi-detector computed tomography: beyond coronary lumenography. Clin Radiol 2007;62(6):518–27.
- Valente AM, Powell AJ. Clinical applications of cardiovascular magnetic resonance in congenital heart disease. Cardiol Clin 2007;25(1):97–110.
- Teien DE, Wendel H, Bjornebrink J, Ekelund L. Evaluation of anatomical obstruction by Doppler echocardiography and magnetic resonance imaging in patients with coarctation of the aorta. Br Heart J 1993;69(4):352–5.
- Campbell M. Natural history of coarctation of the aorta. Br Heart J 1970;32:633–40.
- Reifenstein GH, Levine SA, Gross RE. Coarctation of the aorta: a review of 104 autopsied cases of the “adult type,” 2 years of age or older. Am Heart J 1947;33:146–68.
- Abbot ME. Coarctation of the aorta of the adult type. II. A statistical study and historical retrospect of 200 recorded cases with autopsy, of stenosis or obliteration of the descending arch in subjects above the age of two years. Am Heart J 1928;3:574.
- Cohen M, Fuster V, Steele P, et al. Coarctation of the aorta: long-term follow-up and prediction of outcome after surgical correction. Circulation 1989;80:840–5.
- Oliver JM, Gallego P, Gonzalez A, et al. Risk factors for aortic complications in adults with coarctation of the aorta. J Am Coll Cardiol 2004;44:1641–7.
- Toro-Salazar OH, Steinberger J, Thomas W, Rocchini AP, Carpenter B, Moller JH. Long-term follow-up of patients after coarctation of the aorta repair. Am J Cardiol 2002;89(5):541–7.
- Maron BJ, Humphries JO, Rowe RD, Mellits ED. Prognosis of surgically corrected coarctation of the aorta. A 20-year postoperative appraisal. Circulation 1973;47:119.
- Vriend J, Mulder B. Late complications in patients after repair of aortic coarctation: implications for management. Int J Cardiol 2005;101:399–406.
- Carr J, Amato J, Higgins R. Long-term results of surgical coarctectomy in the adolescent and young adult with 18-year follow-up. Ann Thorac Surg 2005;79:1950–6.
- Crafoord C, Nylin G. Congenital coarctation of the aorta and its surgical treatment. J Thorac Surg 1945;14:347.
- Gross RE. Coarctation of the aorta; surgical treatment of 100 cases. Circulation 1950;1:41–55.
- Barreiro CJ, Ellison TA, Williams JA, et al. Subclavian flap aortoplasty: still a safe, reproducible, and effective treatment for infant coarctation. Eur J Cardiothorac Surg 2007;31(4):649–53.
- BrewerLAIII, Fosburg RG, Mulder GA,Verska JJ. Spinal cord complications fallowing surgery for coarctation of the aorta: a study of 66 cases. J Thorac Cardiovasc Surg 1972;64:368–81.
- Keen G. Spinal cord damage and operations for coarctation of the aorta: etiology, practice, and prospects. Thorax 1987;42:11–18.
- Krieger KH, Sfiencer FC. Is paraplegia after repair of coarctation of the aorta due principally to distal hypotension during aortic cross clamping? Surgery 985;97:2–7.
- Rocchini AP, Rosenthal A, Barger AC, et al. Pathogenesis of paradoxical hypertension after coarctation resection. Circulation 1976;54(3):382–7.
- Choy M, Rocchini AP, Beekman RH, et al. Paradoxical hypertension after repair of coarctation of the aorta in children: balloon angioplasty versus surgical repair. Circulation 1987;75(6):1186–91.
- Singer MI, Rowen M, Dorsey TJ. Transluminal aortic balloon angioplasty for coarctation of the aorta in the newborn. Am Heart J 1982;103:131–2.Chheorta and aortic disease 123
- Inglessis I, Landzberg M. Interventional catheterization in adult congenital heart disease. Circulation 2007;115;1622–33.
- Shaddy RE, Boucek MM, Sturtevant JE, et al. Comparison of angioplasty and surgery for unoperated coarctation of the aorta. Circulation 1993;87:793–9.
- Fawsy ME, Awad M, HassanW, et al. Long-term outcome (up to 15 years) of balloon angioplasty of discrete native coarctation of the aorta in adolescent and adults. J Am Coll Cardiol 2004;43:1062–7.
- Cowley CG, Orsmond GS, Feola P, et al. Long-term, randomized comparison of balloon angioplasty and surgery for native coarctation of the aorta in childhood. Circulation 2005;111:3453–6.
- Forbes TJ, Garekar S, Amin Z, et al. Congenital Cardiovascular Interventional Study Consortium (CCISC). Procedural results and acute complications in stenting native and recurrent coarctation of the aorta in patients over 4 years of age: a multiinstitutional study. Catheter Cardiovasc Interv 2007;70(2):276–85.
- Carr JA. The results of catheter-based therapy compared with surgical repair of adult aortic coarctation. J Am Coll Cardiol 2006;47:1101–7.
- Attenhofer Jost CH, Schaff HV, Connolly HM, et al. Spectrum of reoperations after repair of aortic coarctation: importance of an individualized approach because of coexistent cardiovascular disease. Mayo Clin Proc 2002;77(7):646–53.
- McKellar SH, Schaff HV, Dearani JA, et al. Intermediate-term results of ascendingdescending posterior pericardial bypass of complex aortic coarctation. J Thorac Cardiovasc Surg 2007;133:1504–9.
- Hager A, Kanz S, Kaemerrer H, et al. Coarctation Long-term Assessment (COALA): significance of arterial hypertension in a cohort of 404 patients up to 27 years after surgical repair of isolated coarctation of the aorta, even in the absence of restenosis and prosthetic material. J Thorac Cardiovasc Surg 2007;134:738–45.
- Gardiner HM, Celermajer DS, Sorensen KE, et al. Arterial reactivity is significantly impaired in normotensive young adults after successful repair of aortic coarctation in childhood. Circulation 1994;89:1745.
- Chen S, Donald A, Storry C, et al. Impact of aortic stenting on peripheral vascular function and daytime systolic blood pressure in adult coarctation. Heart 2008;94: 919–24.
- de Divitiis M, Pilla C, Kattenhorn M, et al. Ambulatory blood pressure, left ventricular mass, and conduit artery function late after successful repair of coarctation of the aorta. J Am Coll Cardiol 2003;41:2259–65.
- Simsolo R, Grunfeld B, Gimenez M, et al. Long-term systemic hypertension in children after successful repair of coarctation of the aorta. Am Heart J 1988;115:1268.
- Kimball TR, Reynolds JM, Mays WA, et al. Persistent hyperdynamic cardiovascular state at rest and during exercise in children after successful repair of coarctation of the aorta. J Am Coll Cardiol 1994;24:194.
- Instebo A, Norgard G, Helgheim V, et al. Exercise capacity in young adults with hypertension and systolic blood pressure difference between right arm and leg after repair of coarctation of the aorta. Eur J Appl Physiol 2004;93:116–23.
- Vriend JW, van Montfrans GA, Romkes HH, et al. Relation between exercise induced hypertension and sustained hypertension in adult patients after successful repair of aortic coarctation. J Hypertens 2004;22:501–9.124 Adult Congenital Heart Disease
- Aggoun Y, Szezepanski I, Bonnet D. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events in children. Pediatr Res 2005;58:173–8.
- Connolly HM, Huston J 3rd, Brown RD Jr, et al. Intracranial aneurysms in patients with coarctation of the aorta: a prospective magnetic resonance angiographic study of 100 patients. Mayo Clin Proc 2003;78(12):1491–9.
- von Kodolitsch Y, Aydin M, Koschyk D, et al. Predictors of aneurysmal formation after surgical correction of aortic coarctation. J Am Coll Cardiol 2002;39:617–24.
- Landzberg MJ, Murphy DJ Jr, Davidson WR Jr, et al. Task Force 4: Organization of delivery systems for adults with congenital heart disease. J Am Coll Cardiol 2001;37: 1187–93.
- Graham TP Jr, Driscoll DJ, Gersony WM, et al. Task Force 2: Congenital heart disease. J Am Coll Cardiol 2005;45(8):1326–33.
- Kingwell B, Boutouyrie P. Genetic influences on the arterial wall. Clin Exp Pharmacol Physiol 2007;34:652–7.
- De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62(4):417–26.
- Treasure T. Cardiovascular surgery for Marfan syndrome. Heart 2000;84:674–8.
- Arbustini E, Grasso M, Ansaldi S, et al. Identification of sixty-two novel and twelve known FBN1 mutations in eighty-one unrelated probands with Marfan syndrome and other fibrillinopathies. Hum Mutat 2005;26(5):494.
- Badauy CM, Gomes SS, Sant’Ana Filho M, Chies JA. Ehlers-Danlos syndrome (EDS) type IV: review of the literature. Clin Oral Investig 2007;11(3):183–7.
- Loeys B, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med 2006;355:788–98.
- Williams J, Loeys B, Nwakanma L, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007;83:S757–63.
- Pannu H, Tran-Fadulu V, Milewicz D. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet 2005;139C:10–16.
- Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner’s syndrome in adulthood. Endocr Rev 2002;23(1):120–40.
- Weytjens C, Bove T, van der Niepen P. Aortic dissection and Turner’s syndrome. J Cardiovasc Surg 2000;41:2.
- Loscalzo ML, Van PL, Ho VB, et al. Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics 2005;115(3):732–5.
- Braverman AC, G¨uven H, Beardslee MA, et al. The bicuspid aortic valve. Curr Probl Cardiol 2005;30(9):470–522.
- Cecconi M, Nistri S, Quarti A, et al. Aortic dilatation in patients with bicuspid aortic valve. J Cardiovasc Med (Hagerstown) 2006;7(1):11–20.
- LeMaire SA, Wang X, Wilks JA. Matrix metalloproteinases in ascending aortic aneurysms: bicuspid versus trileaflet aortic valves. J Surg Res 2005;123:40–8.
- Grotenhuis H, Ottenkamp J, Westenberg J, et al. Reduced aortic elasticity and dilatation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol 2007;49:1660–5.
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DV. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004;44:138–43.Chapter 8 Coarctatioaortic disease 125
- Brenner DJ, Hall EJ. Computed tomography: an increasing source of radiation exposure. N Engl J Med 2007;357(22):2277–84.
- Yetman AT. Cardiovascular pharmacotherapy in patients with Marfan syndrome. Am J Cardiovasc Drugs 2007;7(2):117–26.
- Selamet Tierney ES, Feingold B, Printz BF, et al. Beta-blocker therapy does not alter the rate of aortic root dilation in pediatric patients with Marfan syndrome. J Pediatr 2007;150(1):77–82.
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312(5770):117–21.
- Lacro RV, Dietz HC, Wruck LM, et al. Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J 2007;154(4):624–31.
- Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol 2005;45(8):1340–5.
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109(22):2807–16.
- Bonow RO, Cheitlin MD, Crawford MH, Douglas PS. Task Force 3: valvular heart disease. J Am Coll Cardiol 2005;45(8):1334–40.
- Juvonen T, Ergin MA, Galla JD, et al. Prospective study of the natural history of thoracic aortic aneurysms. Ann Thorac Surg 1997;63:1533–45.
- Coady MA, Rizzo JA, Hammond GL, et al. What is the appropriate size criterion for resection of thoracic aortic aneurysms? J Thorac Cardiovasc Surg 1997;113:476–91.
- Vahanian A, Baumgartner H, Bax J, et al. Guidelines on the management of valvular heart disease: The Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology. Eur Heart J 2007;28(2):230–68.
- Bonow RO, Carabello BA, Chatterjee K, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease.Areport of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am
- Coll Cardiol 2006;48:e1–148. [Published erratum in J Am Coll Cardiol 2007;49:1014.] Bentall H, De Bono A. A technique for complete replacement of the ascending aorta. Thorax 1967;23:338–9.
- Gott VL, Pyeritz RE, Magovern GJ Jr, et al. Surgical treatment of aneurysms of the ascending aorta in the Marfan syndrome. Results of composite-graft repair in 50 patients. N Engl J Med 1986;314(17):1070–4.
- Underwood MJ, El Khoury G, Deronck D, et al. The aortic root: structure, function, and surgical reconstruction. Heart 2000;83:376–80.
- Karck M, Kallenbach K, Hagl C, Rhein C, Leyh R, Haverich A. Aortic root surgery in Marfan syndrome: comparison of aortic valve-sparing reimplantation versus composite grafting. J Thorac Cardiovasc Surg 2004;127(2):391–8.
- Kallenbach K, Karck M, Pak D, et al. Decade of aortic valve sparing reimplantation: are we pushing the limits too far? Circulation 2005;112(9 Suppl):I253–9.
Để lại bình luận
Bạn cần phải đăng nhập để đăng bình luận.