Glenn N. Levine, MD, FACC, FAHA
Người dịch: BS. Phạm Trần Xuân Hồng
- Sinh lý cơ bản trong bệnh cơ tim hạn chế là gì?
Sinh lý cơ bản trong bệnh cơ tim hạn chế là sự gia tăng độ cứng của thành tâm thất, gây suy giảm khả năng đổ đầy thất thì tâm trương dẫn đến tình trạng suy tim. Chức năng tâm thu thường bảo tồn (ít nhất là trong giai đoạn đầu của bệnh, mặc dù có thể suy giảm nặng trong giai đoạn sau của bệnh amyloidosis)
- Các nguyên nhân chính của bệnh cơ tim hạn chế là gì?
Khoảng 1/2 các trường hợp bệnh cơ tim hạn chế có nguyên nhân xác định. Nguyên nhân thường gặp nhất có thể xác định được là sự thâm nhiễm cơ tim ở bệnh nhân bị amyloidosis. Những bệnh gây thâm nhiễm khác bao gồm sarcoidosis, bệnh Gaucher, bệnh Hurler. Các bệnh do dự trữ bao gồm hemochromatosis, bệnh dự trữ glycogen, và bệnh Fabry. Nội mạc cơ tim bị ảnh hưởng do xơ hóa nội mạc cơ tim, bức xạ, điều trị anthracycline cũng có thể dẫn đến bệnh cơ tim hạn chế. Mặc dù bệnh cơ tim hạn chế là nguyên nhân tương đối hiếm gây suy tim ở Bắc Mỹ và Châu Âu, nhưng nó là nguyên nhân phổ biến gây suy tim và tử vong ở các vùng nhiệt đới, bao gồm Châu Phi, Trung và Nam Mỹ, Ấn Độ và các vùng khác của Châu Á (nơi tỷ lệ xơ hóa nội mạc cơ tim tương đối cao). Các nguyên nhân của bệnh cơ tim hạn chế được tóm tắt trong Bảng 30-1.
- Những dấu hiệu siêu âm tim thường gặp trong bệnh cơ tim hạn chế là gì?
Trên siêu âm tim chức năng tâm thu bình thường hoặc gần như bình thường, thể tích tâm thất bình thường hoặc giảm, hai nhĩ dãn, độ dày thành tâm thất có thể bình thường hoặc tăng nhẹ, và giảm khả năng đổ đầy thất (rối loạn chức năng tâm trương). Những dấu hiệu trên siêu âm này có thể khác biệt trong giai đoạn cuối bệnh amyloidosis và trong một vài trường hợp khác (xem thêm câu 4).
- Ảnh hưởng của amyloidosis lên tim như thế nào?
Cũng như ở các cơ quan khác, trong bệnh amyloidosis có sự lắng đọng protein vào mô cơ tim. Thuật ngữ amyloidosis được báo cáo lần đầu bởi Virchow và có nghĩa là “dạng bột”. Cơ tim trở nên rắn, dai, và không đàn hồi. Sự lắng đọng protein này dẫn đến các sinh lý bệnh của bệnh cơ tim hạn chế, cũng như các rối loạn chức năng tâm thu sau đó và các bất thường về mặt dẫn truyền. Do đó các bệnh nhân bị ảnh hưởng cả chức năng tâm trương lẫn tâm thu. Họ rất nhạy cảm với dịch, chỉ có ranh giới rất nhỏ giữa quá tải dịch và thiếu dịch. Họ thường biểu hiện bằng hạ huyết áp tư thế và có thể kèm thêm rối loạn dẫn truyền. Tiên lượng nói chung rất xấu.
- Làm thế nào để chẩn đoán bệnh tim amyloidosis?
Nếu bệnh nhân không biết có amyloidosis, bệnh tim do amyloidosis có thể được nghĩ đến qua các triệu chứng và dấu hiệu suy tim, siêu âm tim cho thấy có rối loạn đổ đầy và dày thành tâm thất, có hình ảnh “lấp lánh” trên siêu âm tim (Hình 30-1), và dày thành tâm thất nhưng lại có điện thế thấp trên điện tâm đồ. Nếu cần có thể xác định chẩn đoán bằng sinh thiết.
|
Bảng 30-1: Phân loại các nguyên nhân của bệnh cơ tim hạn chế |
|
Thâm nhiễm
Dự trữ
Nội mạc cơ tim
Bệnh cơ tim không do thâm nhiễm
|
|
Từ Kushwaha S, Fallon JT, Fuster V: Restrictive cardiomyopathy, N Eng J Med 336-267, 1997 |
- Các biểu hiện tim mạch chính của bệnh sarcoidosis?
Sarcoidosis có thể dẫn đến thâm nhiễm các bạch cầu hạt vào cơ tim dẫn đến xơ hóa và hình thành sẹo. Sự thâm nhiễm có thể thành từng mảng trên khắp cơ tim. Sự thâm nhiễm có thể dẫn đến suy tim sung huyết, block nhĩ thất và ngất (do sự thâm nhập vào hệ thống dẫn truyền), và rối loạn nhịp thất (bao gồm ngất và độ tử). Ảnh hưởng lên phổi gồm bao gồm tăng áp phổi và hậu quả đi kèm trên chức năng tim phải.
- Sinh thiết nội mạc cơ tim có hữu ích trong các trường hợp nghi ngờ bệnh cơ tim hạn chế không?
Sinh thiết nội mạc cơ tim được xem là hợp lý trong bệnh cảnh suy tim đi kèm bệnh cơ tim hạn chế không giải thích được nguyên nhân (chỉ định loại IIa, mức độ chứng cứ C, theo khuyến cáo về sinh thiết nội mạc cơ tim của ACC/AHA/ESC. Sinh thiết nội mạc cơ tim có thể cho biết các bất thường đặc hiệu, như amyloidosis hoặc hemochromatosis, hoặc xơ hóa cơ tim và phì đại tế bào cơ tim phù hợp với bệnh cơ tim hạn chế vô căn. Sinh thiết nội mạc cơ tim không nên thực hiện nếu trên CT hoặc cộng hưởng từ (MRI) gợi ý bệnh nhân có co thắt màng ngoài tim hoặc nếu các xét nghiệm ít xâm nhập khác có thể xác định được nguyên nhân bệnh cơ tim hạn chế.
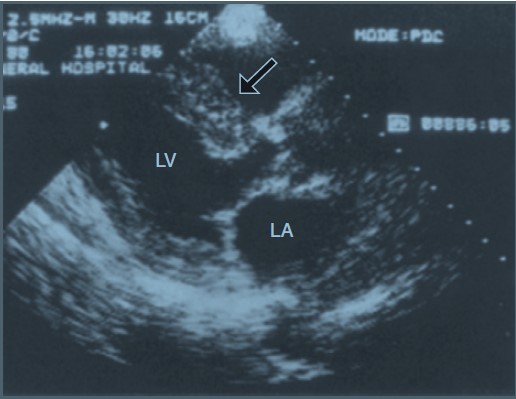
Hình 30-1: Siêu âm tim có hình ảnh lấp lánh ghi nhận được trên bệnh nhân có bệnh tim do nhiễm amyloidosis. Hình ảnh lấp lánh thấy rõ nhất trong hình này ở vách liên thất bị dày.
(Từ Levine RA: Echocardiographic assessment of the cardiomyopathies. In Weyman AE: Principles and practice of echocardiography, ed 2, Philadelphia, 1994, Lea and Febiger, p.810.)
- CT tim hoặc MRI tim có hữu ích trong bệnh cơ tim hạn chế không?
Cả CT tim và MRI tim có thể hữu ích trong bệnh cơ tim hạn chế. Cả 2 phương pháp chẩn đoán hình ảnh này cho thấy màng ngoài tim dày lên, gợi ý co thắt màng ngoài tim là nguyên nhân thực thể của bệnh. MRI tim, với việc sử dụng hình ảnh tăng tín hiệu muộn trong nhiều trường hợp, có thể cho thấy dấu hiệu gợi ý bệnh sarcoidosis, bệnh nội mạc cơ tim nhiễm bạch cầu ái toan, amyloidosis và các nguyên nhân khác của bệnh cơ tim hạn chế. MRI tim có thể được dùng để chẩn đoán bệnh hemochromatosis tim. Trong một số trường hợp, MRI có thể được dùng để hướng dẫn sinh thiết nội mạc cơ tim và đánh giá khả năng lấy được các mẩu sinh thiết để chẩn đoán.
- Hội chứng tăng bạch cầu ái toan (hypereosinophilic – HES) và Loeffler là gi? HES và Loeffler là hai bệnh tương tự nhau (và có thể chỉ là một), đặc trưng bởi tăng bạch cầu ái toan kéo dài mà không có nguyên nhân khác của tăng bạch cầu ái toan (bệnh ký sinh trùng hoặc dị ứng) và gây tổn thương các cơ quan đích qua trung gian bạch cầu ái toan. Biểu hiện tim mạch phổ biến nhất của HES là xơ hóa nội mạc cơ tim, được mô tả đầu tiên vào năm 1930 do Loeffler. Huyết khối thứ phát có thể góp phần gây tắc nghẽn buồng thất, và vì vậy đôi khi người ta gọi đây là “bệnh cơ tim tắc nghẽn hạn chế” (Hình 30-2). Người đọc cần biết rằng có sự trùng lắp và sử dụng lẫn lộn các thuật ngữ HES, bệnh Loeffler, xơ hóa nội mạc cơ tim của Loeffler và xơ hóa nội mạc cơ tim trong y văn, có vài tác giả nghĩ rằng những hội chứng này có liên quan với nhau và một số khác thì điều trị chúng như là những bệnh khác nhau.
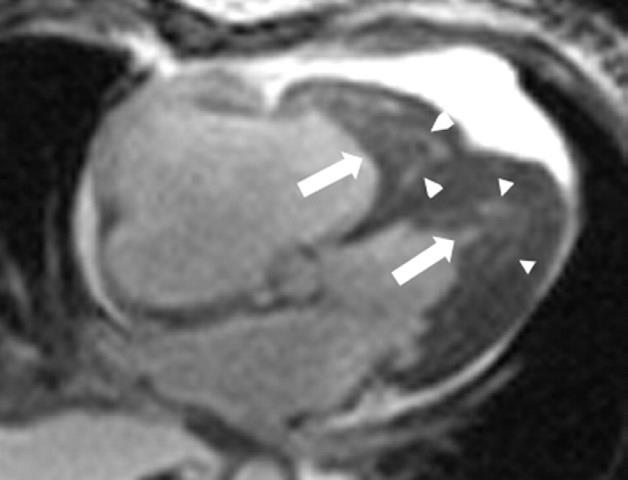
Hình 30-2. MRI tim cho thấy xơ hóa nội mạc cơ tim và huyết khối trong tim, gây tắc nghẽn thất phải và thất trái. Mũi tên dài chỉ huyết khối thất. Mũi tên ngắn chỉ khu vực xơ hóa nội mạc cơ tim.
(Từ Salanitri GC, et al: Endomyocardial fibrosis and intracardiac thrombus occurring in idiopathic hypereosinophilic syndrome. Am J Roentgenol 184:1432-1433, 2005.)
- Bệnh Gaucher là gì?
Bệnh Gaucher là bệnh do sự thiếu hụt enzyme β-glucocerebrosidase, dẫn đến sự tích tụ cerebroside ở tim và các cơ quan khác.
- Hội chứng Hurler ảnh hưởng đến tim như thế nào?
Hội chứng Hurler dẫn đến sự lắng đọng mucopolysaccharide trong cơ tim, van tim, và động mạch vành.
Tài liệu tham khảo, tài liệu nên đọc và websites
- Ammash NM, Tajik AJ: Idiopathic Restrictive Cardiomyopathy: http://www.utdol.com
- Arnold JMO: Restrictive Cardiomyopathy: http://www.merck.com/mmhe
- Cooper LT: Definition and Classification of the Cardiomyopathies: http://www.utdol.com
- Goswami VJ: Cardiomyopathy, Restrictive: http://www.emedicine.com
- Cooper LT, Baughman K, Feldman AM, et al: AHA/ACC/ESC joint scientific statement: the role of endomyocardial biopsy in the management of cardiovascular disease, Circulation 116:2216-2233, 2007.
- Hare JM: The dilated, restrictive, and infiltrative cardiomyopathies. In Libby P, Bonow RO, Mann DL, et al, editors: Braunwald’s heart disease: a textbook of cardiovascular medicine, ed 8, Philadelphia, 2008, Saunders.
- Kushwaha SS, Fallon JT, Fuster V: Restrictive cardiomyopathy. N Engl M Med 335:267-276, 1997.
Để lại bình luận
Bạn cần phải đăng nhập để đăng bình luận.